Analysis
Root mean square deviation (RMSD)
The RMSD of certain atoms in a molecule with respect to a reference structure, rref , is calculated as
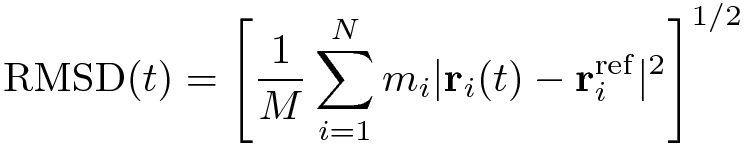
where M = Σi mi and
ri (t) is the position of atom i at time t after least square fitting
the structure to the reference structure.
Usually fitting is done with respect to the starting structure. It is not necessary to use same set of atoms for the RMSD calculation and fitting,
e.g. a protein is usually fitted on the backbone atoms but the
RMSD can be computed for the backbone or for the whole protein.

gmx rms:
Compares two structures by computing the RMSD with each structure in the trajectory (-f) compared with a reference
in the structure file (-s). After typing the command it is
necessary to select an index group for least square fitting and for the RMSD calculation.
We will use the Calpha atoms (Group 3).
gmx rms -f md_protein.xtc -s md_protein.pdb -o rmsd.xvg
Root mean square fluctuation (RMSF)
The RMSF is a measure of the deviation between the position of particle i and some reference position:
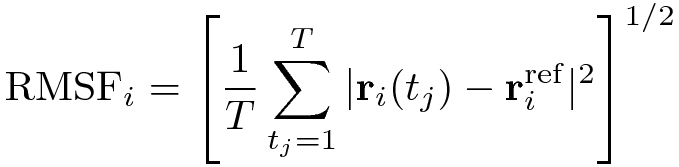
where T is the time over which one wants to average and riref is the reference position of
particle i. This reference position will be the time-averaged position of the same particle i.
The difference between RMSD and RMSF is that the latter is averaged over time, giving a value for each particle i, while for the RMSD the average is
taken over the particles, giving time specific values.
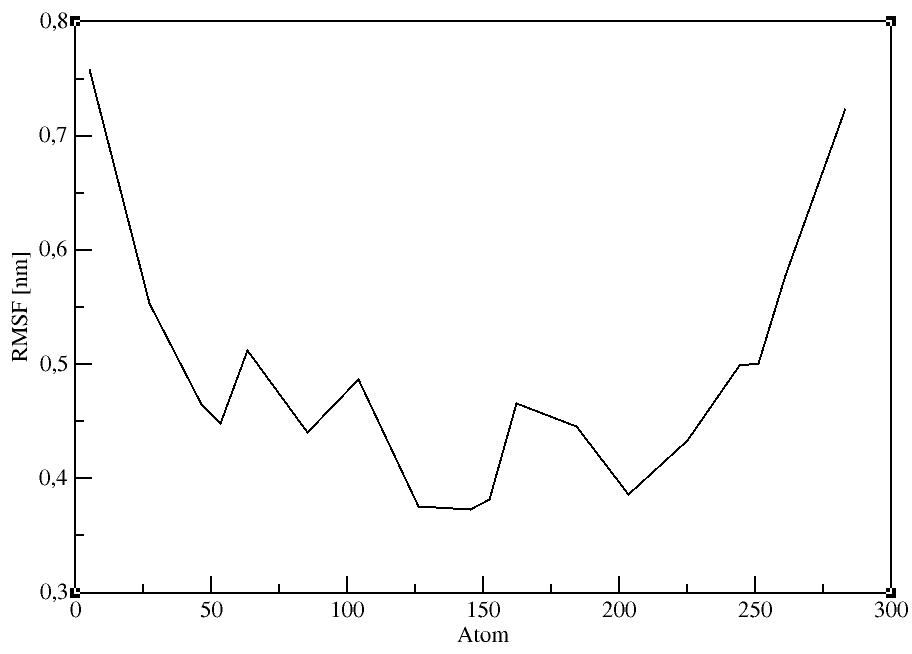
gmx rmsf:
Computes the RMSF (i.e., standard deviation) of atomic positions after (optionally) fitting to a reference frame. A group is selected for the RMSF calculation
after entering the command. We will use the Calpha atoms (Group 3).
gmx rmsf -f md_protein.xtc -s md_protein.pdb -o rmsf.xvg
Radius of gyration (Rgyr)
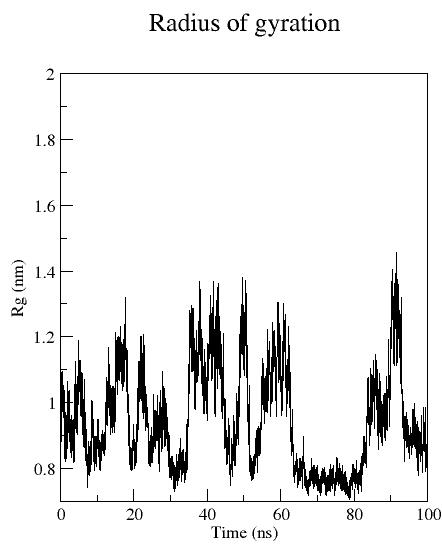
Rgyr is a measure for the compactness of a structure, and calculated according to:
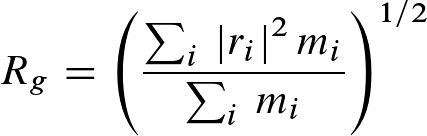
where mi is the mass of atom i and ri
the position of atom i with respect to the center of mass of the molecule.
gmx gyrate:
Computes the radius of gyration of a group of atoms as a function of time.
The atoms are explicitly mass weighted.
gmx gyrate -f md_protein.xtc -s md_protein.pdb -o gyrate.xvg