Setup
This section will explain what is needed to set up an MD simulation with Gromacs, starting with peptide coordinates in a PDB file.
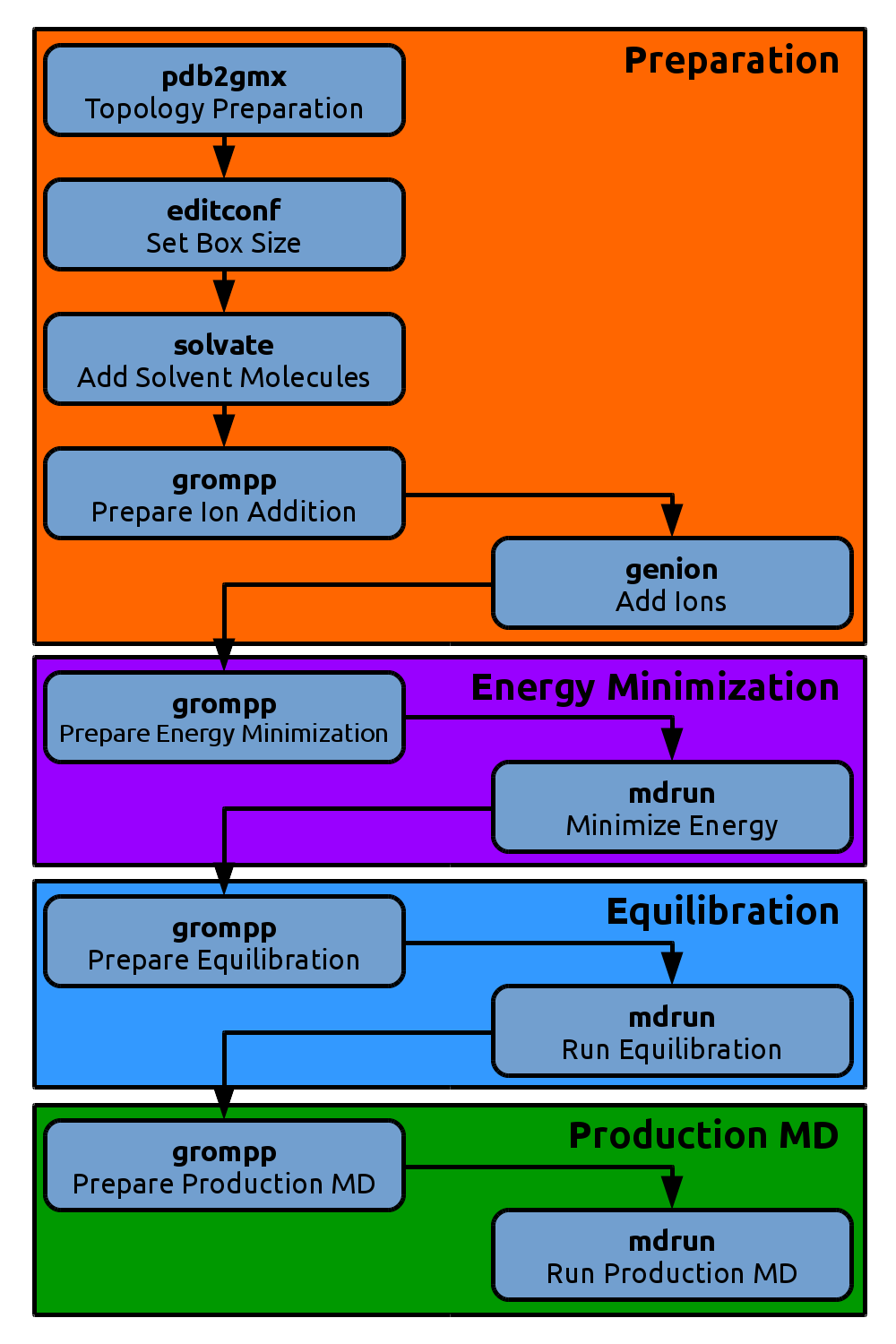
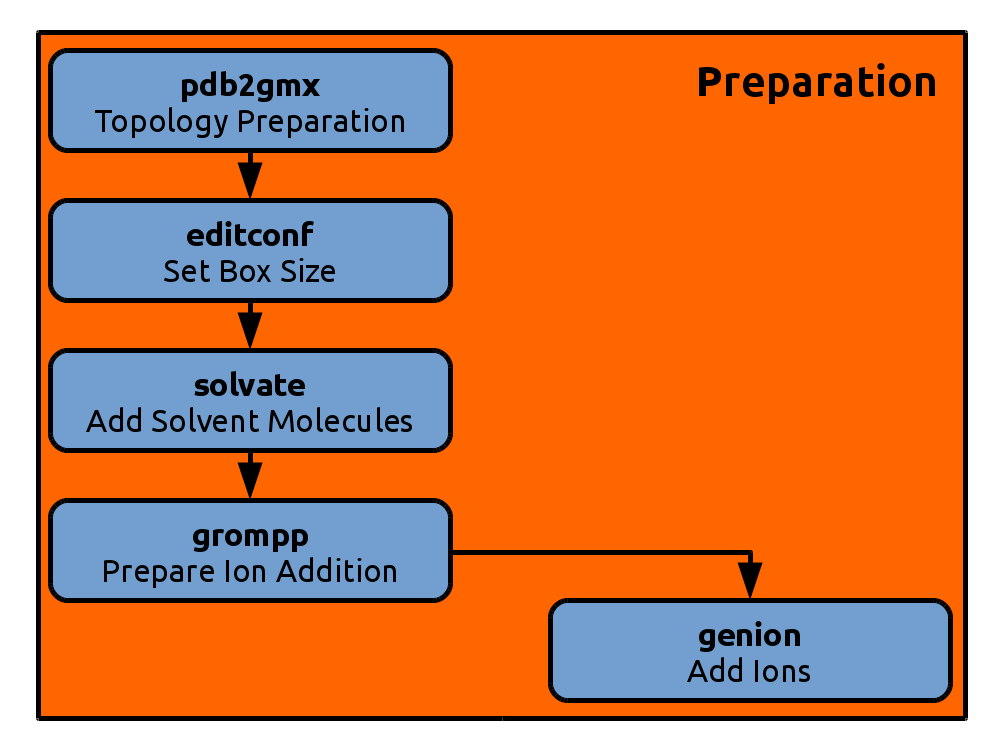
The figures throughout this tutorial give an overview of the current position in the molecular dynamics workflow. They will expand by clicking on them.
Introduction
It takes two ingredients for Gromacs to understand a molecular system: atom
coordinates and topology information. The topology file (typically ending in
*.top) contains information on how many molecules of which kind a system contains,
of what kind their interactions are and the parameters that are used to
compute the energy and forces.
Gromacs can generate topologies for you.
To define a topology, a force field needs to be specified.
A force field is a collection of well balanced parameters for the energy and force computation.
{kind=link}
PDB Formatted Input
Download the file kigaki.pdb from the
Files section
of this tutorial.
Open the file with VMD to have a look at the peptide.
Now generate a topology with the command below. Choose Amber99sb-ildn
as a force field together with the TIP3P water model. While the TIP4P-EW water
model performs better in combination with the Amber force fields, it consists of
four instead of three particles. To limit the simulation size we will use the TIP3P
water model here.
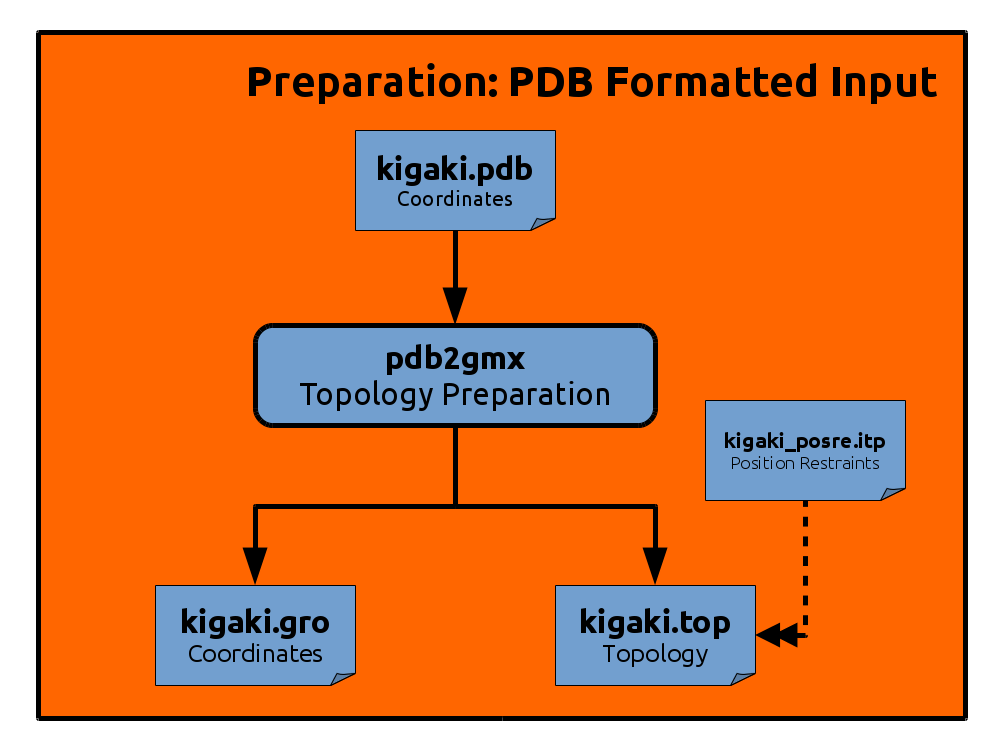
gmx pdb2gmx -f kigaki.pdb -o kigaki.gro -p kigaki.top -i kigaki_posre.itp -ignh
gmx and starts every command.
It will help you to remember that gmx help commands shows you a list of all available
commands with a short description for each of them, while gmx help <command>
gives a more detailed description of the tool whose name you substituded for <command>.
(Gromacs versions before 5.0 consisted of many
programs that have now been merged into gmx.
For compatibility reasons it is therefore still possible to omit gmx and directly start with e.g.
pdb2gmx, but this not guaranteed to work in the future).
The command generated three files. kigaki.gro contains residue and atom names and
the coordinates of all atoms. It could in addition contain velocities for the atoms,
but at this point we haven’t generated any yet.
The second file is the topology file
(kigaki.top). Have a look at its contents using the less command.
After some comments, this file first includes the parameters of the force field in use:
; Include forcefield parameters
#include "amber99sb-ildn.ff/forcefield.itp"
#include) instructs Gromacs to substitute the contents of the
specified file at that particular position in the topology file. The forcefield.itp file
contains the force field parameters of the Amber99sb-ildn force field, which is contained in the Gromacs installation folder.
The next section defines molecule types to make them known to Gromacs.
[ moleculetype ]
; Name nrexcl
Protein 3
The third file we have generated is
kigaki_posre.itp.
It contains position restraints for all heavy atoms of the protein,
i.e. harmonic potentials that couple the atoms to reference positions.
This will be necessary during preparatory steps for the MD simulation to let the water equilibrate around the protein without
the protein itself moving significantly,
so it cannot get deformed by unequilibrated water molecules.
After that, topologies for the water molecules and ions are included as well. Finally the system is given a name and the number of each molecular species in the system is listed:
[ system ]
; Name
Protein in water
[ molecules ]
; Compound #mols
Protein 1
pdb2gmx to optimize hydrogen bonding networks.We have now generated a topological description of our peptide.
Setting the Simulation Box
The next step is to create a box around our peptide that we can later fill with solvent. Gromacs can generate boxes of various shapes: triclinical cells, cubes, dodecahedra and octahedra. The triclinical cell has box vectors at right angles, but with different length, while the cubical box has in addition box vectors of equal length. The solute will be centerd in the simulation box with a minimum distance to any wall of 10 Ångstrom (1.0 nm). As the speed of the simulation decreases approximately linearly with the number of particles, we want the box to be as small as possible, but at the same time keep the solute away from its periodic images at all times. The triclinical cell does not work well, as its rectangular shape is problematic for non-globular proteins that might rotate, unless the center of mass rotation is taken care of. We will choose a dodecahedral cell, as it is close in shape to a sphere and minimizes the volume needed to keep the minimum solute to wall distance:
gmx editconf -f kigaki.gro -o kigaki_box.gro -bt dodecahedron -d 1.0
*.gro file.
(You can navigate to the end of a text file in the less program with the G key.
g brings you back to the top)
The numbers in this final line correspond to the box vector components (in nm) in the order:
v1(x) v2(y) v3(z) v1(y) v1(z) v2(x) v2(z) v3(x) v3(y).
The latter 6 numbers may be omitted for rectangular boxes and Gromacs only supports boxes with
v1(y)=v1(z)=v2(z)=0.
Generate some of the other box shapes too and have a look at them with VMD by loading the corresponding *.gro file and typing
pbc box in the VMD console.
The dodecahedral and octahedral boxes appear not as you might have expected.
The reason is that the boxes are always represented in a general, brick shaped unit cell representation for efficiency.
Consult the section on periodic boundary conditions in the Gromacs manual to get an idea of how the different representations are transformed into each other.
Solvation
Now the box can be filled with solvent molecules.
We have selected the TIP3P water model before, so we need to provide the coordinates of a three-particle water model.
As there are no TIP3P coordinates in the Amber force field directories, we will use the coordinates from spc216.gro.
This file will be found automatically by Gromacs, without specifying a detailed path.
Note that we are still using the correct parameters of the TIP3P water model and only borrowing the coordinates from 216 water molecules,
which were produced with the SPC water model.
gmx solvate -cp kigaki_box.gro -cs spc216.gro -o kigaki_solvated.gro -p kigaki.top
Adding Ions
It is also possible to add some ions in order to mimic simulation conditions with specified salt concentrations.
However, ions are only approximated as charged point particles and do not possess more detailed electronic structure,
which might be relevant for ion binding sites and transition metals ligated to proteins.
Though for NaCl the simple point charge description is usually sufficient.
We add enough ions to neutralize the system and add an additional concentration of 100 mM NaCl.
For doing so, we need an *.mdp file that contains molecular dynamics parameters.
In general, an *.mdp file contains information on the algorithms and options to use during
a simulation with Gromacs.
It is parsed by the Gromacs preprocessor (grompp) and combined with initial coordinates
and velocities into a *.tpr file.
*.tpr files contain the complete information that is needed for an energy minimization or
any kind of MD simulation with gromacs and is the only required input file for
Gromacs' computing engine mdrun.
So why do we need an *.mdp file at this step,
as we only want to add ions to our system?
Gromacs tries to insert the ions at optimal positions that minimize the energy of the system.
Therefore it needs to know which algorithms and parameters to use to compute the energy.
ions.mdp (popup).
ions.mdp:
gmx grompp -f ions.mdp -po ions.out.mdp -c kigaki_solvated.gro -p kigaki.top -o ions.tpr -maxwarn 1
grompp. However, since the system will be
neutralized at the next step, we can ignore this warning which is done with the flag -maxwarn 1.
The resulting *.tpr file will be fed into a tool that acually generates the ions:
gmx genion -s ions.tpr -o kigaki_ions.gro -p kigaki.top -pname NA -nname CL -neutral -conc 0.1
SOL group,
which contains all atoms of the solvent (water) molecules.
The ions will then be substituted for water molecules at positions that minimize the overall electrostatic energy.
Exercises
- Have a look at the resulting system in VMD. The ions can be made visible with the selection keyword ions, using a VdW representation.