Analysis
Solvent accessible surface area (SASA)
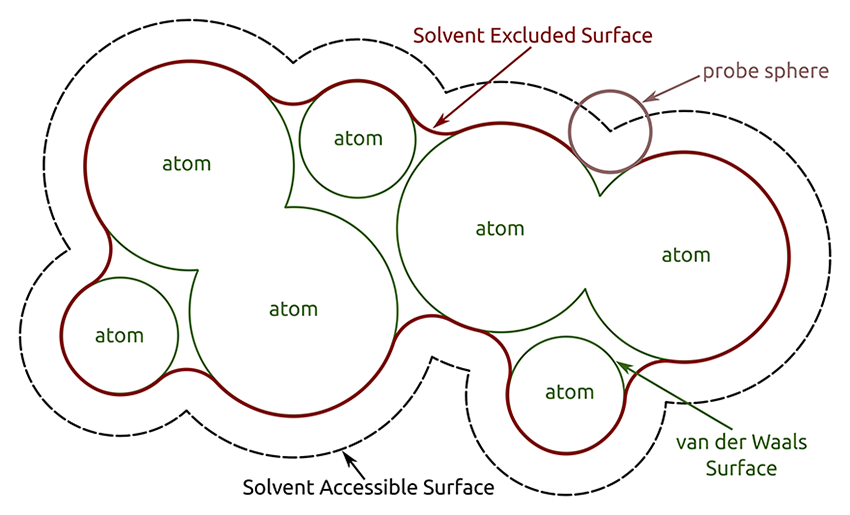
If you would like address the surface properties of your peptide or protein, e.g. whether the surface is rather polar or non-polar, or if you would like to distinguish between exposed and buried areas or amino acids, the calculation of the solvent accessible surface area (SASA) is an easy way to do so. Gromacs offers the sasa tool for this purpose. The calculation can be performed by first defining the radius of a probe sphere (usually the van der Waals (vdW) radius of water) which is 'rolled' along the surface of the selected groups (e.g., the protein). If the probe overlaps with the vdW radii of the atoms of these groups, the area is not accessible by the solvent and therefore accounted to the solvent excluded surface. The closest area around the selected groups without any overlap, which is still accessible by the probe, is the solvent accessible surface area (SASA).
gmx sasa:
Calculates the SASA of the selected groups as a function of time. The user is prompted to interactively select for the surface calculation, which always consist of all non-solvent atoms in the system (e.g. you have a protein in water with NaCl, select the protein). Instead of interactive selections the user can provide the group using the flag -surface. Furthermore, the user may optionally select several subsets of the surface calculation group using the additional flag -output, for which the SASA will be calculated in addition to the SASA of the surface calculation selection. An index file containing the groups for the additional outputs needs to be generated beforehand using the gmx make_ndx tool and is provided using the flag -n.
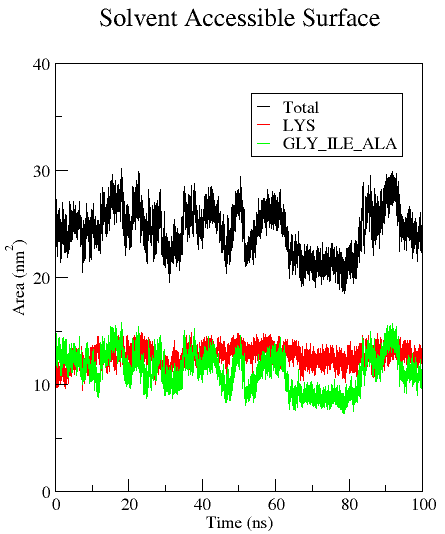
Example: You would like to calculate the SASA of the [KIGAKI]3 peptide, but also of the polar/charged and non-polar residues only. First, the user generates an index file (polar_non-polar_residues.ndx) with a group containing the whole protein (group 1, present by default), a group that includes all the polar and charged residues (Lys for KIGAKI, group 10), and a group containing all the non-polar residues (Ile, Gly, Ala for KIGAKI, group 11). This can be done using gmx make_ndx with the selection 'r Lys' and 'r Ile | r Ala | r Gly' :
gmx make_ndx -f md_protein.tpr -o polar_non-polar_residues.ndx
gmx sasa -f md_protein.xtc -s md_protein.tpr -n polar_non-polar_residues.ndx -o area.xvg -surface 1 -output 10 11 -tu ns
area.xvg provides the time (in ps) by default in the first column, the SASA of the selected groups (in the example: the whole protein) at the respective time in the second column, and the SASAs of the additional outputs in the following columns. The user may calculate additional output files containing e.g. the average area per residue (-or) or the average area per atom (-oa). You can change the unit for time values from the default (ps) to e.g. ns by using -tu ns. For more information regarding the algorithm, see: Eisenhaber F, Lijnzaad P, Argos P, Sander C, & Scharf M (1995) J. Comput. Chem. 16, 273-284.